If your CUT&Tag sequencing failed to meet suggested metrics (Figure 1) or the data are noisy, there are could be several potential reasons. Here, we review troubleshooting recommendations for various challenges encountered during sequencing. Note that our troubleshooting approaches often rely on the use of EpiCypher-recommended control antibodies (H3K4me3, H3K27me3, and IgG) and the SNAP-CUTANA™ K-MetStat Panel, as well as our comprehensive control checks (Figure 1).
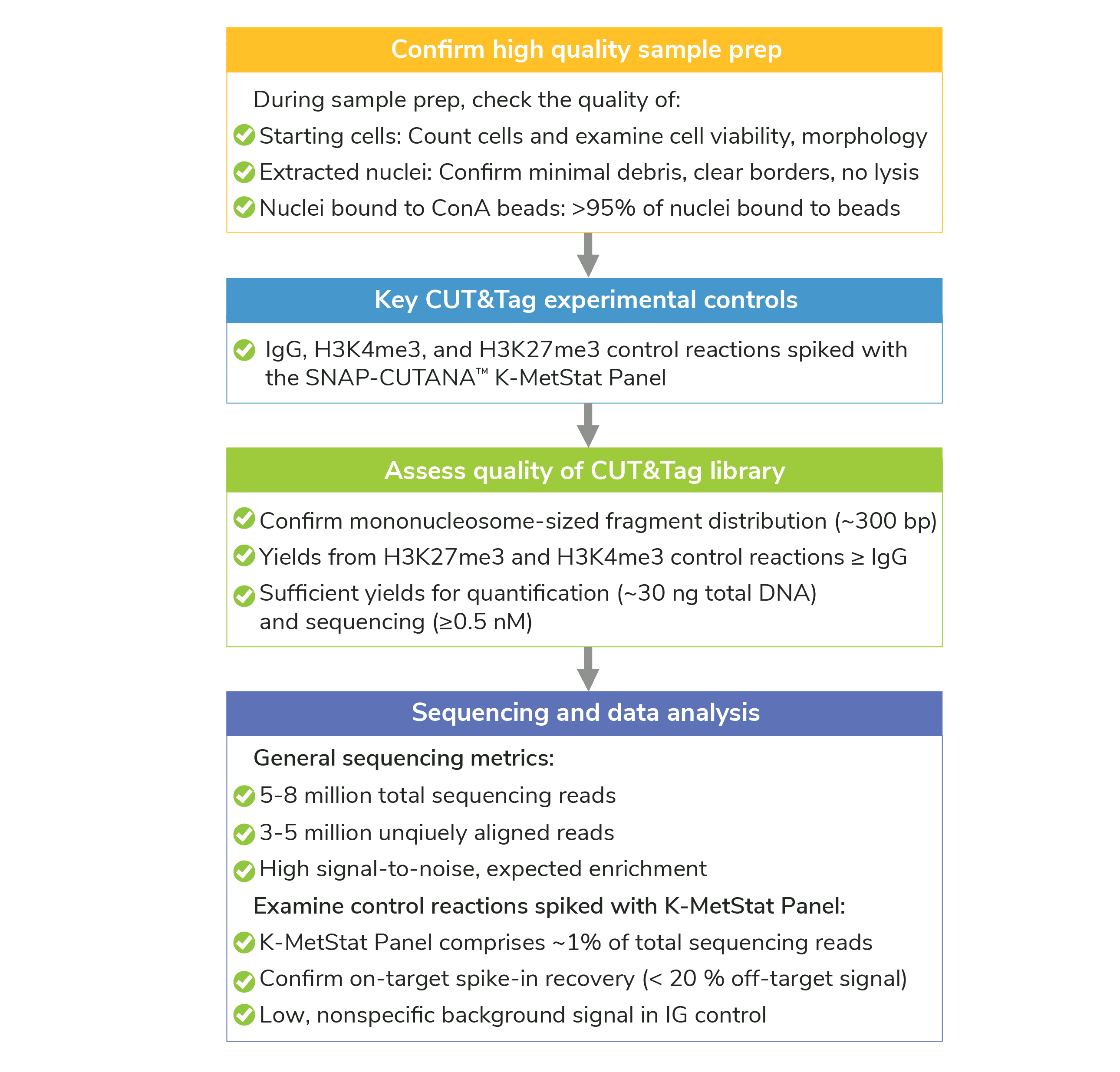
Figure 1: Summary of controls and success metrics for CUT&Tag assays.
Troubleshooting results from a low-concentration CUT&Tag library
Low yields are common when mapping from small numbers of starting cells or if mapping a low abundance target. See our article about troubleshooting low CUT&Tag yields for specific information about improving yields.
In general, we recommend repeating the experiment. If it is not possible to repeat your CUT&Tag experiment, use a Speedvac to increase library concentration and add as much as possible to the sequencing library pool. Deeper sequencing is recommended to capture library diversity.
Signal in IgG negative control and/or signal in open chromatin for repressive targets
Signal in negative control reactions or signal in open chromatin may be caused by the preference of Tn5 for accessible chromatin. The high-salt wash after pAG-Tn5 binding helps reduce nonspecific signal at open chromatin, but some background may be present in the IgG control and when profiling repressive targets.
To help minimize background:
Use freshly harvested, native nuclei.
Always include the IgG negative control for comparison.
Confirm high quality sample prep. Cell/nuclear lysis can increase background in sequencing data.
High read duplication rates
High read duplication rates are common in low-concentration libraries, libraries with low read diversity, or in samples that have been over-sequenced. There are many potential causes, including low target abundance, poor antibody quality, low sample input, incorrect number of PCR cycles, low library yields, and high sequencing depth.
For some low abundance targets and for low nuclei numbers, additional PCR and sequencing is needed to capture diversity. High read duplicates are often a necessary trade-off. Duplicates can be assessed and removed using Picard (broadinstitute.github.io/picard).
If duplication rates are concerning, optimize as follows:
Use 100,000 native nuclei per reaction; include reactions with control antibodies and K-MetStat Panel.
Make sure you are using a CUT&Tag-validated antibody, or are testing multiple antibodies for your target.
Confirm sample prep quality and avoid ConA bead loss.
Optimize PCR: test 14, 16, 18 cycles; aim for >2 ng/µL DNA.
Confirm fragment distribution on the TapeStation/Bioanalyzer.
Sequence to a depth of 5-8 million total reads per reaction.
See Figure 1 for a full summary of all quality control checks.
Examine results:
Confirm that control reactions meet desired CUT&Tag workflow and sequencing metrics. Examine K-MetStat Panel data to validate overall workflow.
For experimental targets:
If yields are high, along with high duplication rates:
Reduce the number of PCR cycles. Use the minimum number of PCR cycles that provides >2 ng/µL DNA.
Make sure you are only sequencing to a depth of 5-8 million total reads per library.
If yields are adequate but duplication rates are high:
Indicates poor antibody and/or mapping a low abundance target - high read duplicates may be a necessary trade-off. Duplicates can be assessed and removed using Picard.